
常见的原发性脑肿瘤是胶质瘤。在组织学上,胶质瘤可能类似于星形胶质细胞、少突胶质细胞或室管膜细胞;因此,根据其形态特征,它们分别被分为星形细胞瘤、少突胶质瘤或室管膜瘤。星形细胞瘤表达胶质纤维酸性蛋白,一种在星形胶质细胞中发现的中间丝,通常被用来帮助将胶质瘤归类为星形细胞瘤。由于星形细胞瘤和少突胶质瘤占胶质瘤的多数,因此我们就这两种类型的胶质瘤作一综述。
胶质瘤发生的危险因素尚不清楚,但职业接触有机溶剂或杀虫剂似乎是一个易感因素。根据对胶质母细胞瘤(GBM)肿瘤中巨细胞病毒(CMV)RNA的检测,也表明巨细胞病毒(CMV)感染可能在某些胶质瘤的病因或进展中起作用。以0~50岁年龄组(8~50岁)为多见。
脑胶质瘤患者的症状取决于脑胶质瘤的解剖部位,可能包括头痛、恶心或呕吐、言语、视觉、听觉或平衡能力的改变、情绪和性格的改变、癫痫发作或抽搐以及记忆缺陷。症状出现的时间范围部分取决于胶质瘤的级别;对于GBM肿瘤,症状的出现通常很快。手术活检是确定肿瘤是否为原发性脑肿瘤及诊断瘤种和级别的必要手段。
根据国际卫生组织(WHO)的标准,胶质瘤肿瘤在组织学上分为I级到IV级。I级肿瘤通常预后良好,更常发生在儿童,而II级肿瘤在组织学检查中以细胞增多为特征:这些II级肿瘤的中位生存期为5-8年。Ⅲ级星形细胞瘤(间变性星形细胞瘤瘤)在组织学检查中表现为细胞过多,以及核异型性和有丝分裂图(见图1)。间变性星形细胞瘤的中位生存期为3年。IV级胶质瘤,也被称为GBMs,在组织学检查中根据高细胞性、核异型性、核分裂图和血管生成和/或坏死的证据进行特征性检查(见图2)。GBM肿瘤患者的中位生存期为12-18个月(5个月、15个月),而老年患者(>60岁)的生存期通常略短于中值。
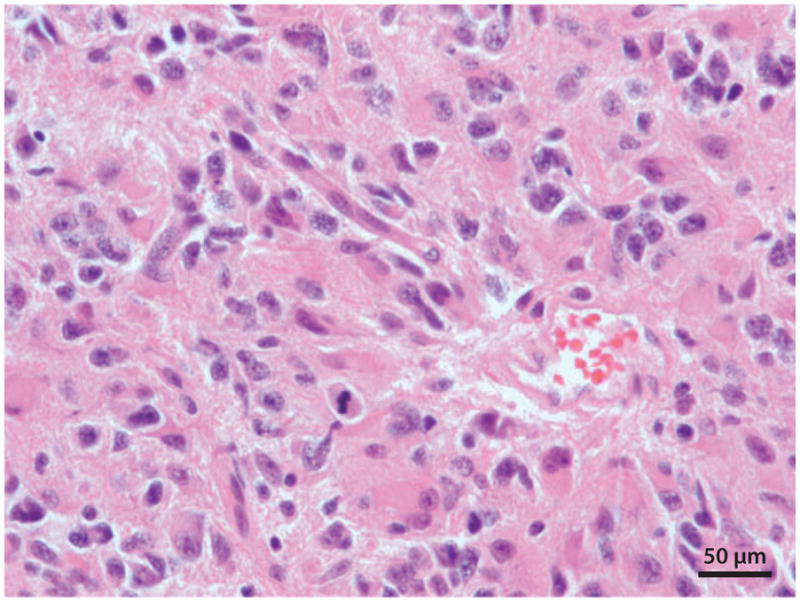
图1:间变性星形细胞瘤(国际卫生组织Ⅲ级)。显微照片的底部中心显示有丝分裂的图形,肿瘤细胞核呈多形性。两者都是典型的间变性星形细胞瘤。
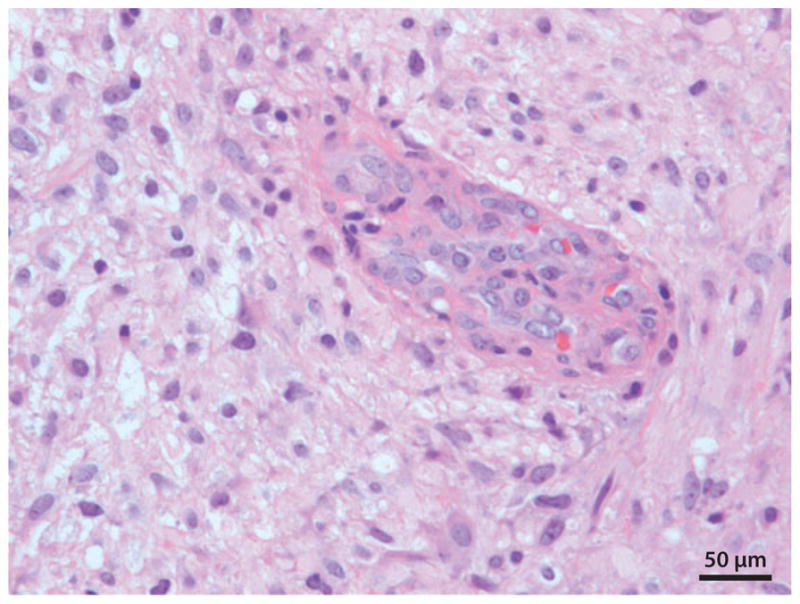
图2:胶质母细胞瘤(国际卫生组织分级IV级)。这张显微照片中央显示内皮细胞增殖(血管生成)。
根据国际卫生组织的标准,少突胶质瘤肿瘤在组织学上分为Ⅱ级和Ⅲ级。在组织学检查中,II级肿瘤表现出高细胞性和淡色的细胞核(见图3),而III级肿瘤(间变性少突胶质瘤)表现出突出的有丝分裂像和血管生成证据的其他组织学特征。
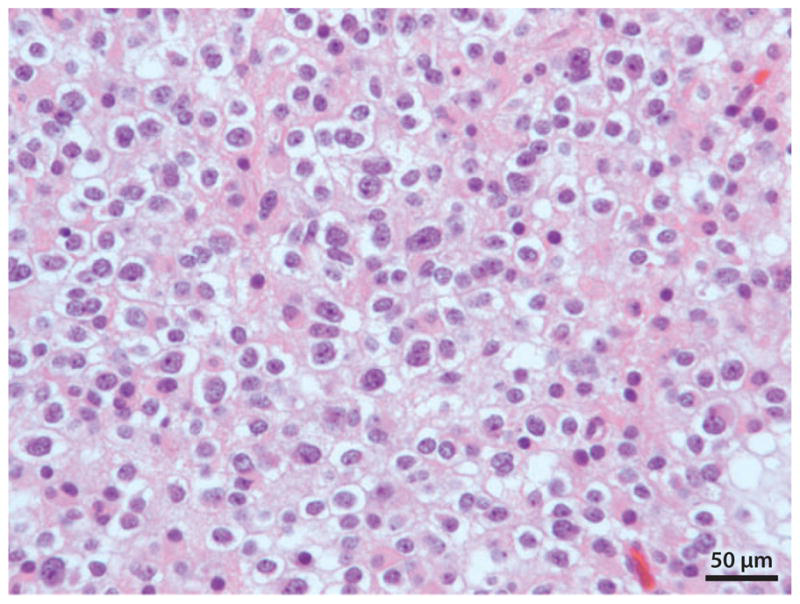
图3:少突胶质细胞瘤(国际卫生组织二级)。这张显微照片显示了典型的少突胶质细胞瘤的清晰的细胞质和单调的单形细胞核。
主要遗传改变
目前对胶质瘤肿瘤细胞基因改变的描述揭示了同一类型和级别的肿瘤之间存在相当大的差异性。这种异质性可能导致目前仅根据胶质瘤类型和级别的组织学分析来评估患者存活率的局限性,并表明根据其遗传表型对某些类型和级别的胶质瘤进行分类将导致更准确地评估生存率和对胶质瘤的反应治疗。
I级肿瘤是良性的,通常不会进展到II、III或IV级肿瘤,其基因改变与II-IV级肿瘤中发现的不同;因此,本文不讨论它们。少突胶质瘤(WHO II级)和间变性少突胶质瘤瘤(WHO III级)经常在1p和19q染色体上表现出杂合度丢失(LOH)(根据研究,在40%-90%的活检中观察到)。这是在少突胶质瘤肿瘤中发现的常见的基因改变,评估对某些化疗药物有良好的反应,对放疗有良好的反应,甚至在复发后也能延长生存期。胶质瘤活检组织可以常规检测1p和19q的LOH,在病理实验室用荧光原位杂交(FISH)或Southern印迹法检测。目前尚不清楚1p和19q位点的哪些基因参与了促进少突胶质瘤肿瘤的生长,也不知道这些基因的丢失如何促进更有利的治疗反应和更有利的预后;然而,这些基因中至少有一个可能参与了少突胶质瘤的发生。少突胶质瘤的另一个常见的基因改变是肿瘤控制因子和脂质磷酸酶PTEN基因的下调。50%的肿瘤中发现了这种基因的下调,这种下调似乎是启动子区域甲基化的结果。血小板衍生生长因子受体α(PDGFRα)扩增发生在约7%的少突胶质瘤。
星形细胞瘤(WHO II级)经常(3%–33%)表现出PDGFRα和/或PDGFRβ基因及其配体PDGF-A和-B或-C和-D的基因扩增。PDGFRα基因的扩增可能是染色体4q12扩增的结果。鉴于神经祖细胞中PDGF-B的逆转录病毒表达可以在新生小鼠和成年大鼠中启动胶质瘤的发生,这些基因改变可能在胶质瘤发生中起重要作用。在不表达高水平PDGF-A和-B的星形细胞瘤中,PDGF-C和-D的表达可能增加,被认为可以替代PDGF-B的促瘤作用。p53缺失也是星形细胞瘤(WHO II级)常见的遗传事件。
在更恶性的星形细胞瘤中,间变性星形细胞瘤(WHO III级)中,约30%的肿瘤检测到编码细胞周期进展调节器Rb的基因丢失,该基因是由于染色体13q13的缺失而发生的。肿瘤控制基因p16INK4A/CDKN2A的下调或突变发生在大约50%的肿瘤中。这种下调可能是由于启动子区域的过度甲基化或染色体9p区域的丢失。p16INK4A和ARF基因由一个称为INK4a/ARF的基因座编码,它位于染色体9p21上,编码p16INK4A和ARF的前体。大约50%的间变性星形细胞瘤肿瘤有p53基因突变。此外,编码内源性p53控制剂MDM2(位于染色体12q)的基因在这些肿瘤中有13%到43%被扩增。由于Rb1/CDK4/p16INK4A和p53/p14ARF基因的改变,负调控细胞周期的信号被中断,导致细胞增殖失控。在大约20%的间变性星形细胞瘤肿瘤样本中也发现了22q号染色体的缺失和7q号染色体的增加,但导致间变性星形细胞瘤肿瘤发生或进展的基因或位点的身份尚不清楚。
GBM肿瘤(WHO IV级)可根据患者的发病年龄和肿瘤的基因改变分为原发性和继发性肿瘤。原发性GBM肿瘤在老年患者(通常>60岁)中一次出现,无早期胶质瘤,约占全部GBM肿瘤的90%。继发性GBM肿瘤起源于已存在的II级或III级星形细胞瘤或混合性胶质瘤(少星形细胞瘤)。在原发性GBM肿瘤中,在7号染色体上发现的编码表皮生长因子受体(EGFR)基因的扩增和/或突变发生在高达60%的肿瘤中。常见的突变是由于外显子2-7的框内缺失导致的功能增益突变;这种突变导致EGFR的组成性激活,从而促进胶质瘤细胞的增殖和侵袭。脂质磷酸酶基因PTEN的缺失,由于染色体10q的缺失或突变,也是原发性GBM肿瘤中常见的遗传现象;这种缺失导致AKT/mTOR活性增加,从而促进细胞存活、增殖和侵袭。无论是EGFR基因的扩增还是PTEN基因的LOH,都可以在病理实验室用FISH或Southern印迹法检测。染色体10q上的其他一些潜在的肿瘤控制基因候选,如DMBT1(在恶性脑肿瘤中删除1)和Myc拮抗剂Mxi1也被提出。此外,MDM2基因(12q号染色体上p53的控制剂)在大约10%到15%的GBM肿瘤样本中被扩增。编码DNA修复酶(MGMT)基因启动子的甲基化在原发性GBM(36%)和继发性GBM(75%)肿瘤中都发生,表明替莫唑胺治疗有更好的疗效。
胶质瘤肿瘤的异质性也存在于单个肿瘤中。例如,胶质瘤肿瘤的某些区域可能会经历缺氧状态。缺氧导致促血管生成基因的激活和血管生成反应的局部增强。此外,血脑屏障的损害可以发生在胶质瘤肿瘤的局部,导致血清来源的细胞外基质蛋白渗漏到肿瘤的某些区域。血清源性细胞外基质蛋白的局灶性表达可改变胶质瘤细胞的整合素信号和运动能力。
增殖和侵袭表型的分子机制
和其他恶性肿瘤一样,胶质瘤也迅速增殖。这种高度增殖的表型是由于多种细胞周期控制剂的丢失,以及来自多个生长因子受体的信号增强,这些受体通过下游效应器发挥对细胞周期调控的作用。在这些肿瘤中启动促增殖信号的生长因子受体包括EGFR和PDGFR。通常,在胶质瘤肿瘤中配体和受体的表达都增加,这表明存在一个自分泌或旁分泌环来放大信号。
重要的是,EGFR和PDGFR生长因子受体与细胞粘附受体(如整合素和ephrins)合作或协调,导致生长因子受体信号的放大。生长因子受体和细胞粘附受体通常能迅速激活粘着斑激酶(FAK),一种胞浆非受体酪氨酸激酶。FAK是细胞周期进程的主要正调节因子,通过增加细胞外信号调节激酶(ERK)活性和cyclind1转录,以及控制p27Kip1的表达而起作用。
胶质瘤是侵袭性肿瘤。对于恶性胶质瘤来说,侵袭性表型是一个高度特征性的特征,其他人把这种表型称为特征性特征。与增殖表型一样,生长因子受体信号在与细胞粘附受体和蛋白酶协同或协同促进侵袭性表型方面发挥着重要作用。多种生长因子受体已被证明能促进胶质瘤细胞的迁移和侵袭,包括c-Met、EGFR和PDGFR。典型的是,肿瘤中生长因子受体和配体的表达增加,再次表明促进信号传导的自分泌或旁分泌环已经到位。
细胞粘附受体的几个不同家族的成员,包括整合素家族的成员,Eph/Ephrin家族的成员和CD44家族的成员,已经被证明可以促进胶质瘤细胞的迁移和侵袭。在某些情况下,细胞粘附受体的表达,如整合素αvβ3和αvβ5,在恶性胶质瘤肿瘤中增加。整合素受体提供与细胞骨架的相互作用,从而产生牵引力,使细胞能够向前推进。关于Eph/Ephrin家族,目前的数据表明Ephrin-B3配体和Eph-B3受体促进胶质瘤细胞侵袭。来自不同种类或家族的细胞表面受体可能以一种环境依赖的方式合作或协调信号事件,这种方式也受到时间上的调控。
胶质瘤细胞中的信号分子作用于细胞表面生长因子受体和细胞粘附受体的下游,放大和传播侵袭前信号。这些信号分子包括细胞质酪氨酸激酶、接合器分子和细胞骨架蛋白。例如,酪氨酸激酶FAK和这个家族的另一个成员Pyk2都可以促进胶质瘤细胞的迁移和侵袭。Src家族酪氨酸激酶也是胶质瘤细胞侵袭所必需的。来自Crk相关底物(CAS)家族的衔接分子,如HEF1和p130CAS,促进胶质瘤的侵袭,并且Crk家族的衔接分子在这个过程中作用于HEF1或CAS蛋白的下游。调节胶质瘤细胞存活和增殖的两个信号分子,磷脂酰肌醇-3激酶(PI3K)和PTEN,也调节胶质瘤细胞的迁移和侵袭。PI3K正调控胶质瘤细胞的迁移和侵袭。PTEN似乎对这些过程负调控;因此,恶性胶质瘤中PTEN功能的丧失可以促进胶质瘤细胞的侵袭。
胶质瘤细胞侵袭很可能需要蛋白酶降解细胞外基质。一些蛋白酶家族,包括丝氨酸蛋白酶、组织蛋白酶、基质金属蛋白酶(MMPs)和ADAMTS金属蛋白酶家族已被证明在胶质瘤细胞迁移和侵袭中起作用。在肿瘤中,蛋白酶活性可由多种因素调节。这种调节的一个重要方面是蛋白酶功能在肿瘤细胞膜的特定区域的定位。这个过程的一个例子是丝氨酸蛋白酶尿激酶的定位。体内GBM肿瘤中尿激酶表达增加,下调尿激酶或其受体(尿激酶受体)控制胶质瘤细胞侵袭。尿激酶与其受体的结合将蛋白酶定位于细胞膜的特定区域,并促进其在这些区域的活性,因为尿激酶与其受体的结合是实现较佳蛋白酶活性所必需的。此外,该受体与细胞膜上的特定整合素受体共定位,进一步指定显示蛋白酶活性的膜区。二个例子是MMP-2与细胞表面整合素αvβ3的结合,它既定位又增强了这种蛋白酶的活性。因此,蛋白酶与细胞表面受体和下游信号分子协同作用,促进胶质瘤细胞侵袭。
恶性星形细胞瘤和少突胶质瘤动物模型的建立
已经建立了星形细胞瘤肿瘤的动物模型。编码抑癌基因p53和Nf1的中枢神经系统特异性失活导致小鼠自发发生Ⅱ级和Ⅲ级星形细胞瘤以及GBM肿瘤。这种胶质瘤的发生可以通过PTEN基因的单倍不足而加速,并且在神经祖细胞中,p53的条件失活与PTEN和Nf1的单倍不足相协调,以诱导星形细胞瘤瘤的形成。这些模型支持这样一个概念,即人类肿瘤中的基因改变,如p53缺失和PTEN功能丧失,可能在星形细胞瘤(Ⅱ级和Ⅲ级)的发生中起重要作用。
GBM肿瘤的啮齿动物模型也可用。在体细胞基因转移模型中,组成活性Ras和Akt的同时逆转录病毒表达导致高级别胶质瘤的形成,其形态与人类GBM肿瘤相似。尽管Ras突变在GBM肿瘤中并不常见,但一项研究表明,由于点突变,人GBM活检中Ras活性增加。在小鼠中,EGFR扩增和p53+CDK4过度表达的缺失或INK4a-ARF的缺失都足以诱导胶质瘤瘤的形成,类似于人类GBM肿瘤(120121)。在EGFR转基因小鼠模型中,p16INK4a、p19ARF和PTEN的LOH与EGFR的扩增协同诱导高浸润性GBM肿瘤。同时,小鼠中枢神经系统中p53和PTEN的同时缺失会产生一种急性发作、高度恶性的胶质瘤肿瘤,其组织学上类似于人类GBM肿瘤。利用PDGF-B在成年大鼠神经祖细胞中的逆转录病毒表达,建立了GBM肿瘤的新模型。在这个模型中,颅内注射含有PDGF-B的逆转录病毒或与PDGFRα联合导致GBM样肿瘤的发生。到目前为止,单个调控细胞周期的基因(如p53、INK4a或ARF)的损害或失活还不足以启动体内胶质瘤的发生。综上所述,这些研究表明神经祖细胞的改变可能至少导致一些高级胶质瘤。
上述模型的使用存在局限性。其中包括肿瘤细胞并非来源于幽默,啮齿动物在某些情况下可能需要几个月才能可靠地发展成胶质瘤。
恶性星形细胞瘤的异种移植模型已被广泛用于评估各种信号分子或基质蛋白在胶质瘤生长和侵袭中的作用。将人类恶性星形细胞瘤/胶质瘤细胞移植到免疫受损小鼠(裸鼠或SCID)的异种移植模型具有相对快速的优点,可用于评估特定分子在体内或消较调节增殖和/或调节侵袭中的作用。人类异种移植模型的一个缺点是大多数人脑胶质瘤细胞系在体内繁殖时不具有侵袭性。另一个缺点是,人类恶性星形细胞瘤/胶质瘤细胞系在培养物中的繁殖会导致关键基因改变的丢失,例如突变型EGFR的表达,这些改变在胶质瘤发生中较为重要。通过在裸鼠(皮下或脑内)而不是在培养中繁殖原发性人类GBM肿瘤,克服了这一局限性;当这些肿瘤在体内繁殖时,患者活检中发现的基因改变得以保留。对于异种移植模型,在原位环境(大脑)中传播肿瘤进行实验分析也很重要,因为大脑中的微环境(即细胞外基质、生长因子和基质细胞)与皮下组织中的微环境不同。
建立了几种少突胶质瘤动物模型。一个模型使用体细胞基因转移技术,其中逆转录病毒表达的PDGF-B被注射到新生小鼠的脑内,导致PDGF-B在神经祖细胞中表达,并诱发少突胶质瘤肿瘤。神经祖细胞或表达异位PDGF-B的星形胶质细胞异种移植也能在12周后诱发小鼠少突胶质瘤肿瘤。这些模型支持这样一个概念,即通过上调PDGF-B配体来上调PDGFR信号足以诱导胶质瘤的发生。
癌症干细胞的贡献
表达神经祖细胞标志物的肿瘤细胞,能够自我更新和分化,被称为癌症干细胞。通常,在一个肿瘤中,这些细胞占肿瘤细胞的比例不到5%,尽管在不同的研究中,这些细胞的数量差异很大。这些细胞的一个关键特征是,与注射非癌症干细胞肿瘤组相比,在免疫缺陷小鼠中注射这些细胞可以更快地形成肿瘤。这些细胞的另一个关键特征是它们能够在免疫缺陷小鼠中形成高度侵袭性的异种移植瘤。这些细胞被认为位于肿瘤的血管周围区域,称为血管龛。通常用于识别这些癌症干细胞的分子包括CD133。胶质瘤中的肿瘤干细胞可能参与了肿瘤的发生、发展为高侵袭性表型和辐射抗性。
结论
胶质瘤中基因和表观改变的特性导致了蛋白质结构-功能研究,阐明了信号通路是如何改变的以及它们对细胞增殖、存活和侵袭的影响。这些研究清楚地表明了调控这些过程的复杂性,并提出了一个动态的过程,即肿瘤细胞通过受体之间的合作和交叉信号通路对其微环境做出环境依赖的反应。它们还表明了肿瘤细胞如何通过重塑其微环境来促进侵袭。重要的是,对这些基因改变的了解使科学家能够创建啮齿动物模型来测试这些改变在体内的重要性,以确定它们对胶质增生或肿瘤进展是否必要,并测试针对这些改变相关通路的新治疗方法。
对神经胶质瘤不同类型和阶段的遗传和表观改变的认识不断增加,已经对诊断产生了影响。由于神经胶质瘤的特征为每个瘤种和级别创建了子类,治疗可能会变得更加个体化,并导致治疗神经胶质瘤的个体化药物方法。目前,对于IV级肿瘤(GBMs),标准的治疗方法是手术减瘤,其次是放疗和化疗。针对肿瘤发生级联的不同方面的几种不同方法正处于不同的发展阶段,其中一些已进入临床试验。然而,动物模型显示,恶性胶质瘤肿瘤对新疗法产生了耐药性;因此,该领域需继续向前发展,以便能够开发出与肿瘤耐药发展步伐相匹配的新的治疗方法和策略。
资料来源:Annu Rev Pathol.2010;5:33–50.doi:10.1146/annurev-pathol-121808-102109
 胶质瘤
胶质瘤 垂体瘤
垂体瘤 脑膜瘤
脑膜瘤 脑血管瘤
脑血管瘤 听神经瘤
听神经瘤 脊索瘤
脊索瘤



 沪公网安备31010902002694号
沪公网安备31010902002694号