
胶质瘤分子病理诊断共识更新背景:脑胶质瘤确诊需通过肿瘤切除或活检获取标本,进行组织病理与分子病理整合诊断,明确病理分级及分子亚型。分子标志物对个体化诊疗及预后评估具有重要价值。脑胶质瘤治疗以手术切除为主,联合放疗、化疗等综合治疗。手术可缓解临床症状、延长生存期,并提供充足标本用于组织病理学与分子病理学诊断。
随着病理学发展与检测技术进步(尤其二代测序、DNA甲基化谱等组学技术),胶质瘤遗传背景及发病机制逐步明确。众多分子标志物被证实对胶质瘤分类、分型、分级、预后及治疗具有关键作用。当前诊断指南中具有诊断价值的基因及染色体改变如下:

2025年6月,中华医学会病理学分会发布《脑胶质瘤分子病理诊断专家共识》,结合国内外研究进展与实践经验,阐述重要分子指标、诊断流程、技术优劣及检测路径,提出适用于中国临床实践的分子整合诊断流程:
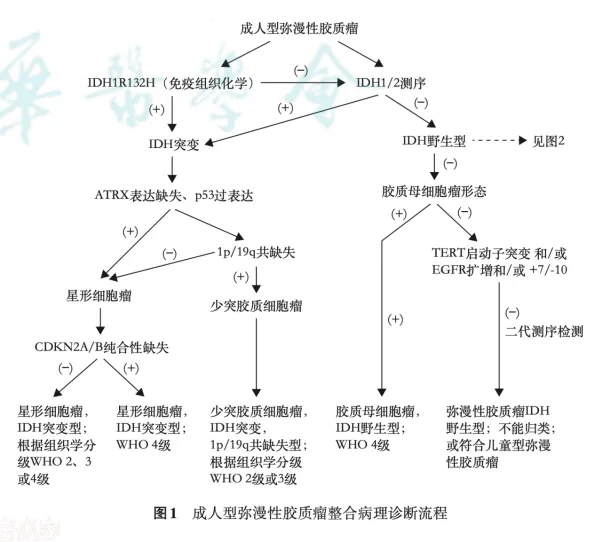
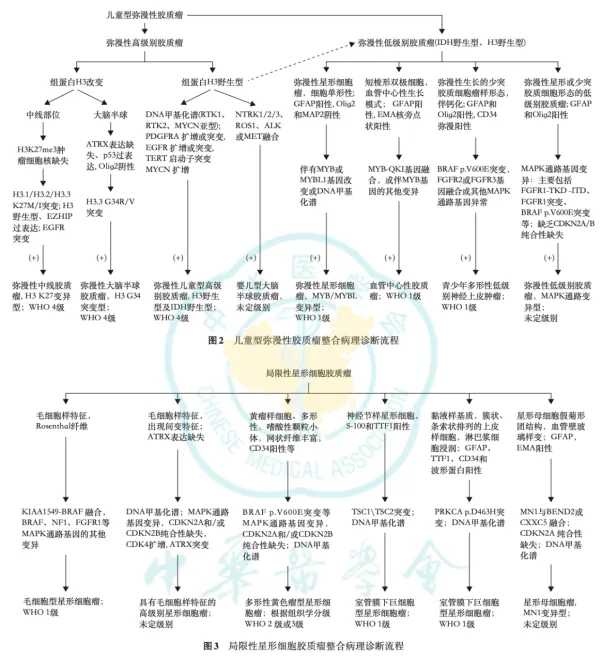
1. IDH1/2 基因突变
IDH基因(异柠檬酸脱氢酶基因)编码三羧酸循环关键酶。胶质瘤相关亚型包括IDH1和IDH2,主要见于IDH突变型星形细胞瘤及IDH突变伴1p/19q共缺失型少突胶质细胞瘤。IDH1突变集中于第132位精氨酸残基(R132)密码子(CGT),IDH2突变集中于第172位精氨酸残基(R172)密码子(AGG)。
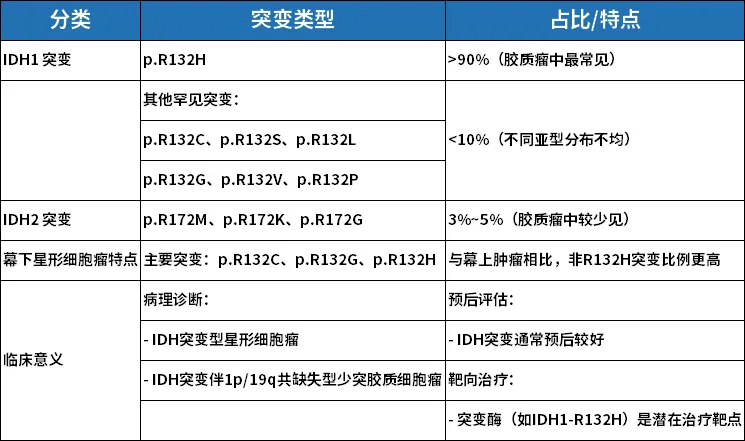
IDH1/2基因突变在胶质瘤中的分布及临床意义
2. 1p/19q共缺失
1p/19q共缺失由1号染色体长臂(1q)与19号染色体短臂(19p)不平衡易位形成融合染色体1q/19p所致,导致单条1号染色体短臂(1p)与单条19号染色体长臂(19q)完全缺失。荧光原位杂交(FISH)仅检测到单拷贝1p和19q,故属杂合性缺失(LOH)。
1p/19q染色体臂不完全或部分缺失不符合少突胶质细胞瘤诊断标准,但可见于IDH野生型胶质母细胞瘤。存在1p/19q共缺失与IDH突变的少突胶质细胞瘤生长缓慢,对PCV(丙卡巴肼+洛莫司汀+长春新碱)联合化疗及替莫唑胺更敏感,总生存期显著延长。
3. TERT基因启动子突变
端粒酶逆转录酶(TERT)通过催化端粒复制维持细胞增殖。TERT启动子突变(主要为C228T或C250T)可持续激活TERT,赋予肿瘤细胞无限增殖能力。
IDH突变型胶质瘤(尤其少突胶质细胞瘤)常伴TERT启动子突变,具辅助诊断价值
成人IDH野生型弥漫性胶质瘤若存在TERT启动子突变,应整合诊断为IDH野生型胶质母细胞瘤(CNS WHO 4级),预后不良
4. CDKN2A/2B基因纯合性缺失
CDKN2A与CDKN2B(位于9号染色体)均为抑癌基因。CDKN2A编码p14^ARF^和p16^INK4a^蛋白,CDKN2B编码p15^INK4b^蛋白,三者均为细胞周期素依赖激酶(CDK)抑制因子,通过抑制CDK活性阻断G1期进程,抑制细胞增殖。CDKN2A/2B纯合性缺失可致细胞增殖失控。
IDH突变且组织学符合2/3级星形细胞瘤者,若存在CDKN2A/B纯合性缺失,整合诊断为CNS WHO 4级IDH突变型星形细胞瘤
CDKN2A未缺失是儿童型MAPK通路改变弥漫性低级别胶质瘤的必要诊断标准之一
5. EGFR基因变异
EGFR基因(位于7p12)编码跨膜酪氨酸激酶受体。
扩增与突变:60% IDH野生型胶质母细胞瘤存在EGFR扩增、突变、重排或剪接变异(扩增最常见)。IDH野生型成人弥漫性胶质瘤若出现EGFR扩增,即使组织学表现为CNS WHO 2-3级且无肾小球样血管增生/假栅栏样坏死,仍整合诊断为IDH野生型胶质母细胞瘤(CNS WHO 4级)
EGFRvIII变异:20%-30%胶质母细胞瘤伴EGFR外显子2-7框内缺失,形成EGFRvIII截短蛋白。其胞内酪氨酸激酶结构域通过二聚化及自磷酸化持续激活下游信号,促进增殖
弥漫性中线胶质瘤突变:多为20号外显子框内插入/复制(编码胞内激酶域),少数为7号外显子错义突变(如p.A289T/V)
6. BRAF基因变异
BRAF编码丝/苏氨酸激酶,介导RAS/RAF/MEK/ERK/MAPK信号通路。胶质瘤中主要为:
BRAF V600E突变:第600位缬氨酸(V)被谷氨酸(E)替换,见于儿童型弥漫性低级别胶质瘤、节细胞胶质瘤、青少年多形性低级别神经上皮肿瘤、毛细胞型星形细胞瘤、多形性黄色瘤型星形细胞瘤及上皮样胶质母细胞瘤
BRAF融合:7q34串联重复致BRAF-KIAA1549融合,多见于毛细胞型星形细胞瘤,亦见于弥漫性软脑膜胶质神经元肿瘤、儿童型弥漫性低级别胶质瘤及局限性星形细胞胶质瘤。罕见融合类型包括BRAF-RNF130、BRAF-CLCN6
7. ATRX基因变异
ATRX基因(X连锁α-地中海贫血/智力低下综合征致病基因)编码ATP依赖解旋酶,参与DNA修复、转录调控、端粒稳定及染色质重塑。第五版WHO CNS肿瘤分类中,ATRX状态为胶质瘤分子分型关键标志。
变异形式:失活变异(突变/缺失/融合),免疫组化显示肿瘤细胞核蛋白缺失(内皮细胞核阳性为内对照)
分布特征:
IDH突变成人弥漫性星形细胞瘤:ATRX失活与TP53突变显著相关
少突胶质细胞瘤:ATRX野生型
亦见于H3K27变异型弥漫性中线胶质瘤、H3G34突变型弥漫性大脑半球胶质瘤及毛细胞样特征高级别星形细胞瘤
8. H3 K27突变
H3K27变异型弥漫性中线胶质瘤(DMG)特征性改变为H3 p.K28M/I(K27M/I)突变,导致组蛋白3第28位赖氨酸残基(K28)三甲基化(me3)障碍,表现为H3K28me3(H3K27me3)广泛缺失。
分子机制:
H3.3/H3.2/H3.1 p.K28M(K27M)突变使K28被甲硫氨酸(M)取代,直接阻断甲基化
K28M突变蛋白抑制PRC2复合物活性(通过结合EZH2),间接导致H3K28me3缺失
致病途径:H3K28me3缺失改变染色质构象,促进癌基因转录/抑制抑癌基因转录
罕见变异:H3 p.K28I(K27I)突变(赖氨酸被异亮氨酸取代)具有相似致病机制
9. H3 G34突变
H3G34突变型弥漫性大脑半球胶质瘤(H3G34 DHG)为大脑半球弥漫浸润性胶质瘤,特征性改变为H3.3(H3F3A)p.G35(G34)R/V突变(第35位甘氨酸(G)被精氨酸(R)或缬氨酸(V)取代)。
分子机制:
阻碍H3.3与SETD2/KDM2A结合
影响组蛋白3第37位赖氨酸[H3 p.K37(K36)]甲基化状态
阻断H3 p.K37(K36)与MutSα错配修复蛋白相互作用
致病途径:共同抑制H3 p.K37(K36)三甲基化(me3),下调H3K37me3(H3K36me3)可上调原癌基因MYCN表达,驱动H3G34 DHG发生
 胶质瘤
胶质瘤 垂体瘤
垂体瘤 脑膜瘤
脑膜瘤 脑血管瘤
脑血管瘤 听神经瘤
听神经瘤 脊索瘤
脊索瘤



 沪公网安备31010902002694号
沪公网安备31010902002694号