
髓母细胞瘤属于儿童期最为常见的恶性脑肿瘤类型之一。一项聚焦于遗传易感性的开创性研究,共纳入了超过1000名髓母细胞瘤患者,Waszak等研究者报告显示,约6%的患者存在与疾病相关的种系改变。在这些存在种系改变的患者中,仅41%表现出相关临床特征,而具有家族史的患者比例为46%,这一数据凸显了单凭临床线索来诊断癌症易感综合征存在局限性。此外,该项研究及其后续工作,进一步明确了与髓母细胞瘤不同分子亚型相关联的新型癌症易感综合征。
回顾以往内容,关于“儿童脑瘤是否与父母基因相关”的探讨指出,几种常见脑肿瘤各自存在“偏好性”的遗传易感基因。世界卫生组织专家委员会曾依据分子标记物,将髓母细胞瘤划分为四个分子亚型,分别为WNT活化型、SHH活化型、Group 3型以及Group 4型。INC国际小儿神经外科专家James T. Rutka教授在其研究工作中指出,髓母细胞瘤的这四种亚型具备不同的临床病理特征,且生存率存在显著差异。
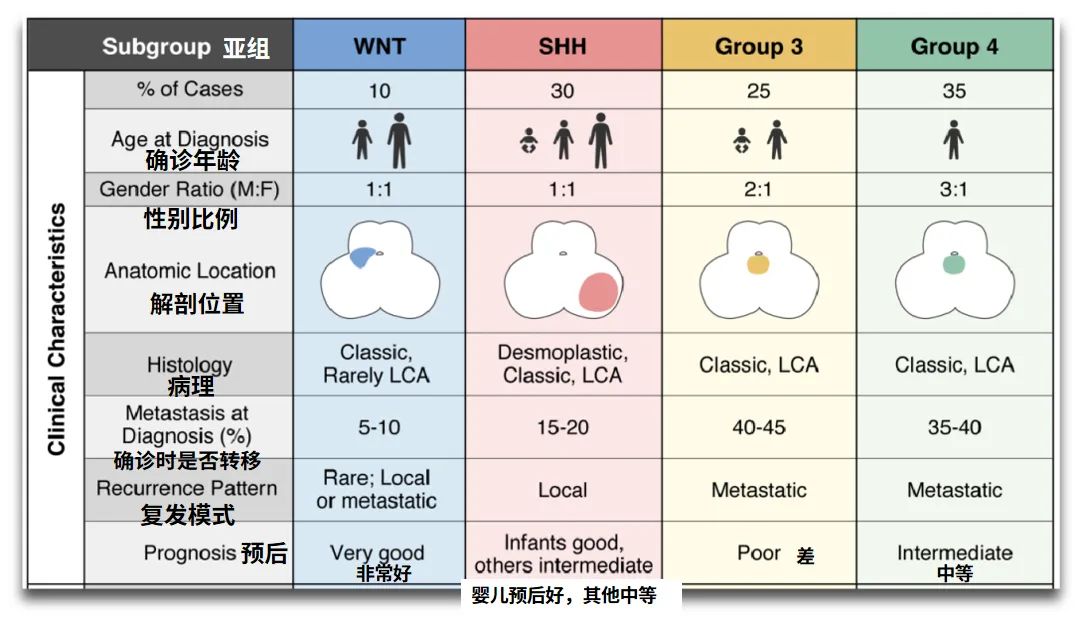
不同分子分型对应的遗传易感性基因亦存在差异。遗传易感性指的是,由于遗传结构存在差异,不同人群或个体在环境因素作用下,表现出易于罹患肿瘤等多基因疾病的倾向。简而言之,遗传的并非肿瘤本身,而是指个体具有“更易患肿瘤”的体质。
01 WNT亚组
在所有WNT亚型髓母细胞瘤患者群体中,存在种系抑癌基因APC功能缺失性致病性变异的比例高达6%至8%。对于不携带体细胞CTNNB1突变的患者而言,这一比例更是超过70%,这类情况常导致常染色体显性家族性腺瘤性息肉病(FAP)综合征的诊断。因此,针对缺乏体细胞CTNNB1突变(或无法进行体细胞检测)的WNT亚型髓母细胞瘤患者,以及患有多发性息肉或具有FAP相关肿瘤个人史或家族史的患者,通常建议进行种系APC基因检测。
鉴于FAP患者发生髓母细胞瘤的发病率相对较低,且预后通常较好,目前并不推荐对此类患者实施常规的神经系统影像学筛查。法国协作组报告指出,虽然FAP相关的WNT亚型髓母细胞瘤患者生存率极佳,但其发生第二肿瘤的风险却很高,这些第二肿瘤包括韧带样纤维瘤、甲状腺癌、毛发基质瘤、骨瘤、血管瘤以及恶性外周神经鞘瘤等,其中许多与治疗(放疗和/或手术)相关。这一发现支持对FAP相关的WNT亚型髓母细胞瘤采取与散发病例类似的降阶梯治疗策略。
在理解息肉病综合征与WNT亚型髓母细胞瘤之间关联的同时,需要注意到,曾有一例成年WNT亚型髓母细胞瘤患者被报道携带与MUTYH相关息肉病相符的种系双等位基因MUTYH致病性变异。然而,目前对于MUTYH相关息肉病综合征患者,尚未制定基于年龄的监测建议。
02 SHH亚组
在SHH亚型髓母细胞瘤中,癌症易感综合征的比例相对较高,现有数据显示其患病率可高达40%。在罹患SHH亚型髓母细胞瘤的婴儿及幼儿中,约有20%患有常染色体显性痣样基底细胞癌综合征(NBCCS,历史上称为Gorlin综合征),该综合征主要由PTCH1(最为常见)或SUFU基因的杂合性种系致病性/可能致病性变异所引起。迄今为止,PTCH2基因尚未被证实是相关的易感基因。
NBCCS的特征在于良性与恶性肿瘤风险均有所增加。据估计,携带SUFU致病性/可能致病性变异的个体,发生SHH亚型髓母细胞瘤的绝对风险在7%至9.2%之间;而对于PTCH1携带者,该风险则为0.37%至1.1%。非中枢神经系统病变包括牙源性角化囊肿(常见于PTCH1携带者,但在SUFU携带者中尚未有描述),此类囊肿可导致严重不适但罕见恶变。此外,患者还可能发生纤维瘤,常见部位包括心脏和卵巢。最为重要的是,受累个体会发生基底细胞癌,其患病率随年龄增长而上升,且在放疗后或过度紫外线照射后也会增加。
由于大多数与NBCCS相关的髓母细胞瘤患者为婴儿,一线治疗通常不包含放疗。对于年龄较大的患者,及时识别潜在的癌症易感综合征,对于指导讨论计划中对颅脊轴进行放疗的必要性及剂量设定至关重要。NBCCS患者中脑膜瘤的发病率有所增加,这包括原发性脑膜瘤以及放疗后继发性脑膜瘤,且在SUFU突变背景下比在PTCH1突变背景下更为常见。
被诊断为NBCCS的个体应接受关于防晒以及避免不必要电离辐射的咨询。例如,在条件允许的情况下,PTCH1致病性变异携带者的年度口腔全景X线片检查可考虑用磁共振成像替代。监测指南需根据所涉及的具体基因进行定制。建议对所有女性患者进行皮肤病学监测、心脏纤维瘤的超声心动图检查以及卵巢纤维瘤的超声检查。对于种系PTCH1携带者,还需额外关注颌骨囊肿的检测。
对于PTCH1和SUFU致病性/可能致病性变异携带者,对髓母细胞瘤的监测主要通过临床手段进行,包括频繁的神经系统检查、头围评估,并对神经系统症状保持高度警惕。目前,仅建议对SUFU携带者在生命的前五年进行常规脑部成像筛查,部分共识指南建议在生命早期将监测间隔缩短至每3个月一次。目前,仅建议对成年SUFU致病性/可能致病性变异携带者,或曾接受过放疗的个体进行脑膜瘤筛查。
已有报道显示,高达5%的婴儿SHH亚型髓母细胞瘤患者存在种系G蛋白偶联受体161基因致病性/可能致病性变异。这些患者可能表现出重叠的NBCCS样表型。其髓母细胞瘤的特征在于体细胞1号染色体q臂存在拷贝中性杂合性缺失。由于目前认为种系GPR161变异导致髓母细胞瘤的绝对风险较低,可能无需进行脑部成像监测。虽然不推荐常规监测,但在与家庭充分讨论数据有限性以及该策略的获益与风险后,可在特定情况下考虑个性化监测策略。最为重要的是,父母应了解髓母细胞瘤可能出现的体征与症状,以便能够及时就诊评估。
近期在年龄较大(中位年龄6.3岁)的儿童中发现,延伸因子复合体基因的双等位基因种系致病性/可能致病性变异,占SHH亚型髓母细胞瘤的14%至21%。这些肿瘤表现出体细胞9号染色体缺失,从而导致ELP1和PTCH1基因的杂合性丢失,以及对侧PTCH1等位基因的双等位基因失活。已有家庭成员罹患髓母细胞瘤的报道,并且近期研究描述了免疫组化在识别受累患者中的应用。
尽管需要对这类患者进行更全面的表征,但目前认为ELP1种系致病性/可能致病性变异携带者发生髓母细胞瘤的风险似乎较低。目前不推荐进行脑部成像监测。然而,如上文关于GPR161监测所述,在与家庭充分讨论数据有限性以及个性化策略的获益与风险后,可在特定情况下予以考虑。提高临床警惕性并对家庭成员进行关于髓母细胞瘤体征和症状的教育可能是必要的,特别是对于髓母细胞瘤患者的杂合子兄弟姐妹。
Li-Fraumeni综合征是一种特征极为明确的癌症易感综合征,患者存在发生包括髓母细胞瘤在内的多种脑肿瘤的风险。在5至16岁的SHH亚型髓母细胞瘤患者中,检测到20%存在种系TP53致病性变异,其中仅4%具有提示LFS的家族史。另一方面,在SHH/TP53髓母细胞瘤患者中,56%的致病性变异携带者患有LFS。这类患者的生存率极差,但放疗对于髓母细胞瘤的生存仍然至关重要。在髓母细胞瘤治疗期间及之后,对LFS患者的监测应遵循相应指南,因为这对其生存具有影响,并且接受放疗者的风险意识应予以提高。
一部分TP53突变的SHH亚型髓母细胞瘤可出现在其他种系综合征背景下,例如与DNA复制修复缺陷相关的综合征,包括体质性错配修复缺陷和聚合酶校对缺陷。这些肿瘤具有独特的免疫生物学特征,包括高突变和基因组微卫星不稳定性,并且适用于免疫检查点抑制剂治疗。这类患者存在罹患血液系统、结肠及其他恶性肿瘤的风险。相关监测建议可参考本系列关于体质性错配修复缺陷的综述。
一部分SHH亚型髓母细胞瘤可发生在由同源重组修复缺陷引起的范可尼贫血背景下。在种系HRD背景下发生髓母细胞瘤的罕见个体,通常发病年龄非常早,往往在FA/HRD诊断之前。在接受癌症治疗期间,这些患者对放疗和化疗(特别是基于烷化剂的化疗)相关毒性高度敏感。因此,对于任何患有髓母细胞瘤并伴有皮肤、骨骼或神经系统异常,或对化疗出现严重、意外毒性的患者,应讨论潜在HRD综合征的可能性。目前不推荐对FA患者进行中枢神经系统成像筛查。具有遗传性乳腺癌和卵巢癌综合征癌症相关家族史,或肿瘤测序显示存在提示HRD的致病性变异或特征性突变谱,可能促使临床考虑进行种系分析,特别是针对髓母细胞瘤患者的BRCA2和PALB2突变。杂合性BRCA2和PALB2变异在SHH亚型髓母细胞瘤患者中富集。然而,鉴于携带单个BRCA2或PALB2致病性/可能致病性变异的个体,其发生髓母细胞瘤的估计风险推测较低,目前不推荐进行专门的神经影像学监测。典型成年发病癌症的杂合子携带者的管理方案,将在未来的AACR癌症易感工作组指南中进行讨论。
03 第3/4组
与WNT和SHH亚组不同,第3组和第4组髓母细胞瘤与已知癌症易感综合征的关联,不如前两者明确。如上文所述,已有关于杂合性BRCA2和PALB2种系致病性/可能致病性变异患者的报道。曾有一名患有Rubinstein-Taybi综合征和种系CREBBP致病性变异的患者被发现患有第3组髓母细胞瘤,但这不足以支持两者存在真正的关联,也无法据此提出监测建议。目前,对第3/4组髓母细胞瘤患者进行癌症易感综合征评估,可仅限于那些有BRCA相关癌症家族史,或肿瘤测序显示存在HRD突变特征的患者。
本文资料来源:Update on cancer predisposition syndromes and surveillance guidelines for childhood brain tumors. Clin Cancer Res. Author manuscript; available in PMC: 2024 Dec 3.
 胶质瘤
胶质瘤 垂体瘤
垂体瘤 脑膜瘤
脑膜瘤 脑血管瘤
脑血管瘤 听神经瘤
听神经瘤 脊索瘤
脊索瘤



 沪公网安备31010902002694号
沪公网安备31010902002694号