
双胞胎兄弟先后确诊胶质瘤的病例引发对疾病遗传性的关注。首例患者为17岁男性,足球运动时突发癫痫就诊,神经系统检查未见异常。MRI影像显示左侧额叶前部存在非强化占位病变。首次手术实现次全切除,病理确诊为WHO 2级少突胶质细胞瘤,伴有1p/19q共缺失。

术后24个月替莫唑胺辅助化疗期间,肿瘤出现进展并发生间变转化,升级为WHO 3级间变性少突胶质细胞瘤。4年后,其21岁兄长同样以新发癫痫起病,MRI检查发现左侧额叶前部4.0×3.1cm病灶,病理证实为具有相同遗传标记的WHO 2级少突胶质细胞瘤。
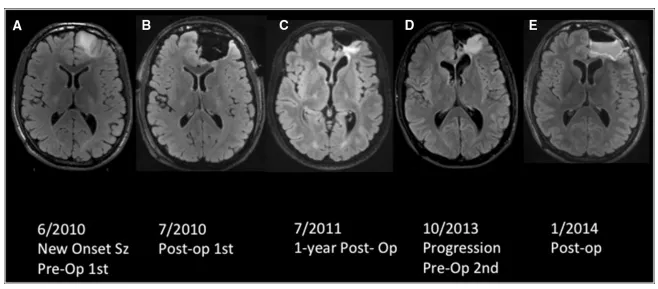
两例患者临床表现具有高度相似性:发病年龄相近,病理亚型一致,肿瘤定位相同,均呈现恶性进展特征。这种镜像式发病模式为家族性胶质瘤遗传学研究提供重要案例。
诊疗过程与病情进展
首例患者完成替莫唑胺12个月周期治疗后,MRI显示T2高信号区域扩大,提示肿瘤进展。继续追加12个周期替莫唑胺治疗后,影像学出现对比增强区域,表明肿瘤发生间变转化。二次手术病理确诊为WHO 3级间变性少突胶质细胞瘤,遗传学检测显示1p19q共缺失、PTEN野生型、无EGFR扩增。术后接受放疗联合CCNU方案化疗,目前病情稳定。
其兄长在次全切除术后3年随访中,MRI显示肿瘤进展接受二次手术,病理证实转化为WHO 3级间变性少突胶质细胞瘤,后续计划接受放化疗联合治疗方案。
遗传学分析与家族史调查
家族史调查显示两例患者为同一家庭中仅有的兄弟成员。母系为俄罗斯裔,父系为英国裔。祖父患有骨髓增生异常综合征,祖母50岁时确诊乳腺癌。父亲结肠镜检查结果阴性。家族中未发现林奇综合征、李-佛美尼综合征或遗传性乳腺癌卵巢癌综合征病例。
肿瘤组织遗传学检测显示,两位患者MLH1、MSH2、MSH6和PMS2错配修复蛋白表达均正常,表明DNA错配修复系统功能完整。目前尚未发现明确的遗传模式。
讨论与临床意义
大多数胶质瘤病例表现为散发性。家族性胶质瘤虽有报道但极为罕见。某些家族性癌症综合征与常染色体显性遗传的胶质瘤风险相关,通常表现为不完全外显率。当两名同胞患病且无已知显性遗传综合征时,需考虑常染色体隐性遗传疾病可能性。
该病例中所有肿瘤均表现为少突胶质细胞瘤病理特征,且在组织学和解剖定位上呈现高度一致性。高外显率家族突变与低外显率易感位点可能共同影响家族性胶质瘤发病风险。这些案例表明,在未发现孟德尔遗传病的胶质瘤家族中,可能存在潜在遗传性病因,需进一步研究明确其遗传学关联。


文献来源:Joseph A. Osorio, et al. Familial gliomas: cases in two pairs of brothers. Journal of Neuro-Oncology, 2014.
 胶质瘤
胶质瘤 垂体瘤
垂体瘤 脑膜瘤
脑膜瘤 脑血管瘤
脑血管瘤 听神经瘤
听神经瘤 脊索瘤
脊索瘤



 沪公网安备31010902002694号
沪公网安备31010902002694号