
女孩脑瘤奇迹般消失医生也无法解释原因……
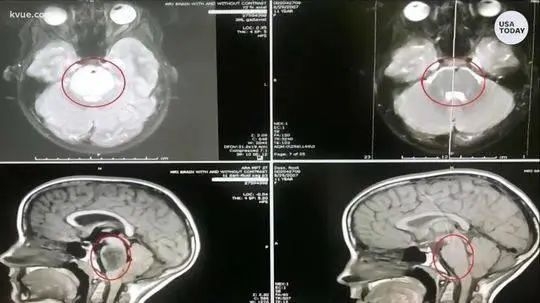
扫描对比图
据今日美国12月18日报道,美国德克萨斯州海斯县一名女孩患脑瘤,且无法手术,但近日,女孩脑中的脑瘤奇迹般的消失了,医生也无法解释原因。现在,德克萨斯州11岁女孩Roxli Doss正在做她喜欢做的事,那就是骑马。她的父亲Scott Doss说“她和以前一样活泼”。
很难想象,就在今年6月Roxli被诊断出患有脑干肿瘤(DIPG),而且无法进行手术。
德克萨斯州奥斯汀戴尔儿童医疗中心的Virginia Harrod医生说:“这种疾病少见,但我们确实发现了它,这是一种致命疾病。会导致吞咽能力下降,或者视力下降,说话能力下降,较终会导致呼吸困难。”
Harrod医生说这名11岁的女孩接受了数周的放射治疗,尽管目前还没有治愈方法。8月,Roxli的家人还为她举行了一次慈善活动,社区作出了很大的支持。
Roxli的父母所能做的就是祈祷奇迹出现,“感谢上帝,我们等到了。”Roxli的父母喜较而泣地说。
Harrod医生说:“当我一开始看到MRI(核磁共振扫描)时,我也觉得难以置信,MRI检测不到肿瘤了,这真的很不寻常。”
Roxli的父亲说“多家医院和多名医生都确认这确实是DIPG”,但医生们也无法解释肿瘤消失的原因。
从无药可治到无迹可寻,这家人说现在他们只有感谢上帝了。Roxli的父母说医生对她的扫描进行了反复检查,只是为了确认结果,Roxli还将继续接受治疗,如免疫治疗,作为预防措施。Roxli的父亲说:“我们不知道她会健康多久,看看她,她做得很棒。”
儿童“肿瘤”——脑瘤
癌症是儿童四大死亡原因,2014年的报告每10个死亡儿童约有一人是死于癌症,在美国其中的四分之一为脑癌,大约为534;在过去的数十年里,统计数据显示“血癌”——白血病是儿童癌症的主要类型。近年来,随着化疗、放疗、骨髓移植的进步,多数白血病可以治愈。
美国疾病预防控制中心(CDC)数据揭示:自2014年开始,脑瘤正式取代白血病成为新的儿童肿瘤,脑瘤成为一多发儿童实体瘤,成为儿童一杀手。
与“退居二线”的儿童白血病相比,儿童脑瘤的“杀伤力”更大。2022年5月19日,《新英格兰医学杂志》(NEJM)发表综述《儿童脑肿瘤》,重点讨论基于分子生物学证据的儿童脑肿瘤较新分类,以及其中较具代表性的胶质瘤和CNS胚胎性肿瘤。本文在此简介其主要内容。

儿童脑肿瘤的分类
2021年发布的五版国际卫生组织(WHO)中枢神经系统肿瘤分类(CNS5)对脑肿瘤分类法作出变更,强调了分子诊断特征。这样就产生了包含分子生物标志物和传统分类(组织学、超微结构和免疫组化特征)的混合命名法,这些变更反映了基于遗传特征确定诊断类别的趋势,遗传特征在许多情况下会驱动预后并提示潜在治疗靶点。
胶质瘤
1、儿童型弥漫性低级别胶质瘤
低级别胶质瘤是儿童较常见的脑肿瘤,如果将混合型胶质神经元和神经元肿瘤也涵盖在内,低级别胶质瘤将占全部病例的1/3(图1A)。这组肿瘤具有异质性;与成人低级别胶质瘤不同,儿童低级别胶质瘤很少转化为更高级别肿瘤。成人低级别胶质瘤(可转化为更高级别肿瘤)中常见的IDH1或IDH2突变体在儿童肿瘤中要少见得多。
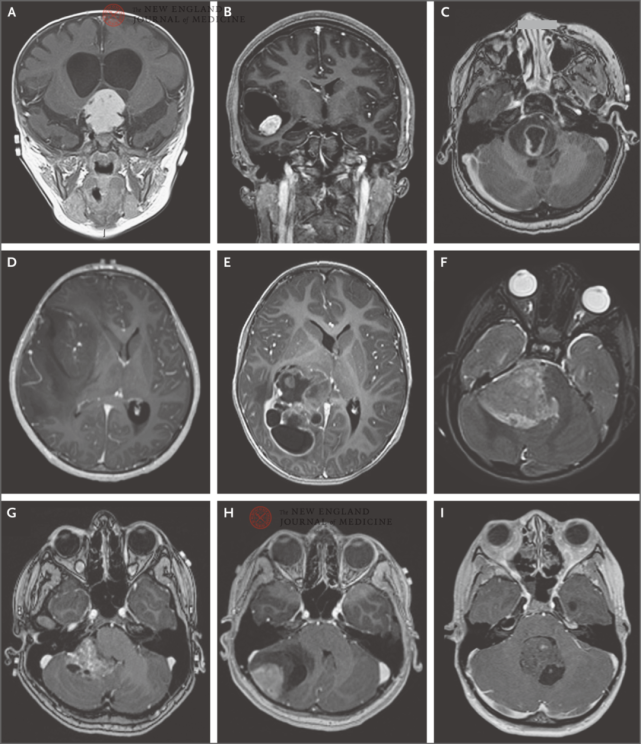
▲图1.儿童典型脑肿瘤的T1加权增强磁共振成像(MRI)扫描
MRI扫描显示出儿童脑肿瘤在部位、大小、强化和内部结构方面的异质性。
大多数儿童低级别胶质瘤的初始治疗是手术,旨在建立组织诊断并较大水平实现顺利切除。在一项大规模国际性研究中,低级别胶质瘤患儿的5年无进展生存率为69%,总生存率为95%。进展的危险因素是低龄、切除不完全、纤维性组织学特征及下丘脑或视交叉部位。
放疗对复发或残留的低级别胶质瘤合适,5年无进展生存率为71%,总生存率为93%。因年龄、解剖部位和遗传特征而有肿瘤进展风险的患儿常采用辅助化疗,原因是担心放疗对发育期的脑部产生神经毒性。
目前已采用可能比传统化疗疗效更好、毒性更低的药物靶向分子改变。研究已经发现BRAF的体细胞改变或NF1的生殖细胞系改变可能在肿瘤发生中起作用。一些低级别胶质瘤发生了BRAF改变,BRAF编码MAPK通路的下游调节因子丝氨酸-苏氨酸激酶蛋白(BRAF)。两种常见的BRAF改变是BRAF V600E癌基因的点突变及BRAF与另一个未知功能的大基因KIAA1549的融合(表1)。

▲表1.儿童低级别胶质瘤中的BRAF癌基因改变
2、毛细胞型星形细胞瘤
较常见的儿童星形细胞瘤是毛细胞型星形细胞瘤,约占儿童、青少年和年轻成人(<20岁)脑肿瘤的20%(图1B)。它们通常生长缓慢且范围局限,10年生存率超过90%。这些肿瘤大多位于小脑和鞍上区,但也可出现在其他部位。尽管毛细胞型星形细胞瘤很少发生恶变,并且通常预后良好,但仍有20%结局不良,出现局部复发或播散。KIAA1549–BRAF融合发生于80%~90%的毛细胞型星形细胞瘤,是颅后窝部位的肿瘤,并且可能与总生存期延长相关。
3、儿童型弥漫性高级别胶质瘤
儿童型高级别胶质瘤占儿童脑肿瘤的10%,预后不良(表2)。儿童型弥漫性高级别胶质瘤的标准辅助治疗是局部姑息性放疗,但长期生存率很低,而且结局在过去50年间并无明显好转。高级别胶质瘤患儿的3年无事件生存率和总生存率分别为10%和20%。脑桥弥漫性中线胶质瘤的结局较差,不接受放疗的情况下,中位生存期为4个月,接受放疗的情况下,中位生存期也只有8~11个月。一般而言,化疗对儿童高级别胶质瘤的效果有限。
目前在研的其他疗法包括免疫检查点控制剂、嵌合抗原受体T细胞疗法、恶性肿瘤疫苗和溶瘤病毒疗法。
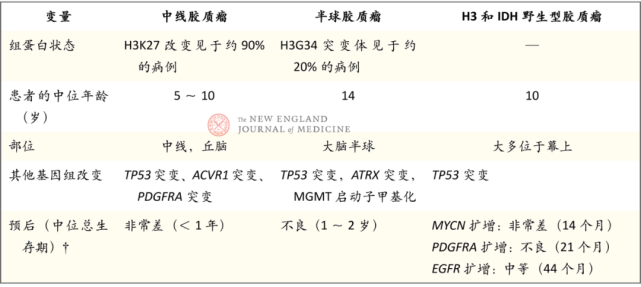
▲表2.儿童型弥漫性高级别胶质瘤
目前已发现胶质瘤的4种亚型。
一种亚型是弥漫性中线胶质瘤,这是一种致死性较高的肿瘤,患病人群为幼童,并且不可切除。新术语“H3K27-改变”取代了之前的术语“H3K27突变”,因为已发现了其他分子改变。对弥漫性中线胶质瘤进行的甲基化研究已发现组蛋白H3中的致癌性组蛋白错义点突变。这些肿瘤患者的生存期比野生型肿瘤患者要差(图1C)。H3K27改变可以比组织学分级更好地评估预后。H3K27改变似乎是儿童弥漫性中线高级别胶质瘤的特异性改变。
二种亚型是弥漫性半球胶质瘤,H3G34-突变型,该亚型起源于较年长儿童和年轻成人的大脑半球。该肿瘤还与其他基因改变相关,包括α-地中海贫血X连锁(ATRX)和TP53肿瘤蛋白53(TP53)突变,以及O6-甲基鸟嘌呤DNA甲基转移酶(MGMT)启动子甲基化。组蛋白突变见于80%以上的中线高级别胶质瘤,以及40%以上的大脑半球胶质瘤,主要发生于儿童。
三种亚型是弥漫性儿童型高级别胶质瘤,H3野生型和IDH野生型(图1D)。这是一种侵袭性肿瘤,通常见于大脑半球,预后不良。
四种亚型是新生儿和婴儿中一种具有临床独特性的肿瘤,称为婴儿型半球胶质瘤,该肿瘤通常携带受体酪氨酸激酶基因融合,包括ALK、NTRK1/2/3、ROS1和MET4。这些激酶改变有可能成为治疗靶点,初步研究提示,激酶融合阳性肿瘤患者的结局好转。
室管膜瘤
室管膜瘤是继胶质瘤和髓母细胞瘤之后,三常见的儿童脑肿瘤,占儿童CNS肿瘤的5%~10%;90%发生于颅内,大多数起源于颅后窝,其余发生于脊髓。室管膜瘤是一组异质性肿瘤,根据组织学特征、分子特征和部位进行分类,至少有9种分子亚型。
室管膜瘤根据间变程度分为1级、2级或3级。少见的室管膜下室管膜瘤为1级。2级和3级室管膜瘤位于幕上或幕下(表3)。幕上室管膜瘤根据两种致癌性分子融合进行分类。C11orf95-RELA融合发生于70%的病例,这一融合导致核因子κB信号通路出现组成型激活(图1E)。C11orf95基因的新命名为ZFTA;ZFTA可与更多配体融合,而不仅仅是RELA。另外一个融合涉及YAP1。据报道,与YAP1融合相比,新命名的ZFTA融合可以比组织学分类更好地评估临床病程,并导致更差预后。
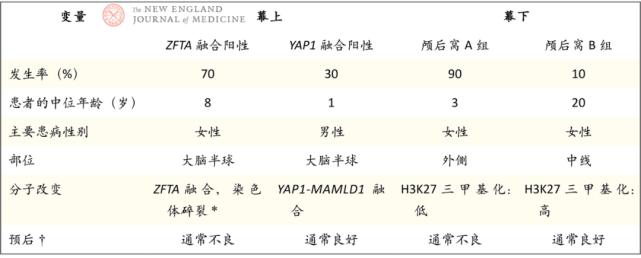
▲表3.儿童室管膜瘤
颅后窝室管膜瘤根据甲基化组学(methylomic)特征细分成两种较常见的亚型:PFA和PFB室管膜瘤。前者主要发生于婴儿,位于外侧,预后比PFB室管膜瘤差。与PFB肿瘤相比,PFA肿瘤存在H3K27三甲基化表观遗传标志物的相对缺失。PFB亚型发生于较年长儿童,一般预后较好(图1F)。然而,当PFA组和PFB组患儿接受适形放疗时,未发现预后价值。
非转移性室管膜瘤患儿首先接受较大限度顺利切除术,之后接受局部适形放疗,婴儿除外。化疗的作用尚未确定,但正在研究中。尽管手术和放疗取得了进展,但儿童室管膜瘤的远期结局仍然不良,10年总生存率和无进展生存率分别为50%和30%。
髓母细胞瘤
低级别胶质瘤是较常见的儿童脑肿瘤,而髓母细胞瘤是较常见的儿童恶性脑肿瘤。髓母细胞瘤通常起源于小脑,患者表现为颅内压增高或小脑功能障碍。髓母细胞瘤占儿童胚胎性肿瘤的60%以上,70%发生于10岁以下儿童,且患病男童多于女童,但年龄和性别差异因肿瘤亚型而异。1/3的病例发生于3岁以下儿童。
与髓母细胞瘤患儿结局不良相关的因素包括:体积大、就诊时为播散性疾病、年龄小(<3岁)以及术后影像学检查显示残留肿瘤大于1.5 cm。
CNS5系统如今包含两种类型的髓母细胞瘤:分子定义的髓母细胞瘤和组织学定义的髓母细胞瘤。分子定义的髓母细胞瘤包含4个亚型,每个亚型具有独特的甲基组学和转录组学特征,以及独特的临床行为(表4)。遗传分析已确定了亚型的亚类,并提出了新治疗策略。
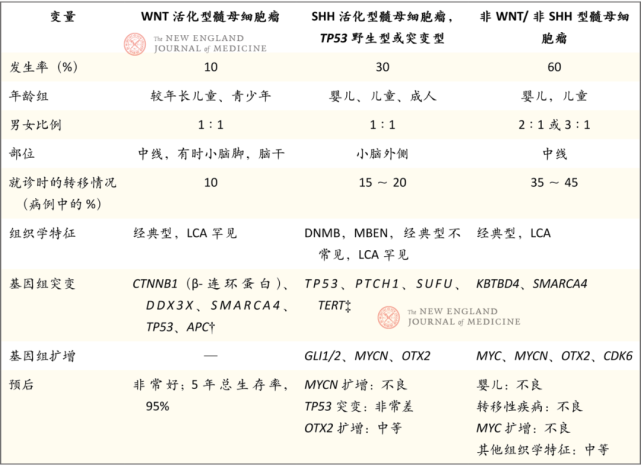
▲表4.分子定义的髓母细胞瘤*
WNT活化型髓母细胞瘤
无翼/集成信号通路(WNT)活化亚型占全部髓母细胞瘤的10%,患病男童和女童比例相同,发生于较年长儿童或青少年(图1G)。肿瘤位于小脑中线,有时累及小脑脚和脑干。WNT型髓母细胞瘤具有典型的组织学特征,通常与CTNNB1编码的β-连环蛋白累积相关。CTNNB1突变见于90%的病例,并导致核β-连环蛋白积累,而后者会促进肿瘤发生。
这些肿瘤的预后好,10年无事件生存率超过95%。它们具有由过量突变β-连环蛋白驱动的异常通透性血管系统,这损害了血脑屏障,有可能可以采用化疗。WNT型肿瘤的这一特征可以解释为什么有些患者发生出血。因为WNT型肿瘤患者的生存期良好,所以目前正在研究减少放疗和化疗的治疗策略。
SHH活化型髓母细胞瘤
音猬因子(SHH)活化型髓母细胞瘤占髓母细胞瘤的30%,两种性别均衡分布,发生于幼童和年轻成人(图1H)。SHH型髓母细胞瘤可根据TP53肿瘤控制基因的存在或缺失情况进行分层。TP53突变(发生于9%的病例)是肿瘤发生的驱动因素,预示着预后不良,而WNT型肿瘤中的TP53突变不影响结局。SHH型髓母细胞瘤的分子分层有助于我们对靶向疗法开展临床试验。一个例子是应用新型SMO控制剂vismodegib和sonidegib治疗难治性或复发性SHH型髓母细胞瘤。
非WNT/非SHH活化型髓母细胞瘤
在目前的命名法中,非WNT/非SHH亚型包括3亚型和4亚型髓母细胞瘤。不同于WNT型和SHH型髓母细胞瘤,上述肿瘤的患病男童多于女童,并且就诊时很有可能已发生转移。它们位于小脑中线,通常具有经典型或大细胞/间变型组织学特征。潜在驱动突变尚未确定(图1I)。
3亚型肿瘤占髓母细胞瘤的25%,发生于婴儿和儿童,预后较差,5年总生存率为50%。细胞遗传学异常常见,包括见于近一半病例的等臂染色体(镜像)17q。对于幼童(<3岁),手术切除后可给予新型疗法,包括大剂量化疗联合自体干细胞挽救疗法以及根据风险采用的其他方案,目的是延迟放疗,避免清髓。
4亚型肿瘤较为常见,占全部髓母细胞瘤的35%。它们发生于较年长儿童和青少年,预后中等,5年总生存率为70%。基因改变包括MYCN癌基因(6%的病例)和CDK6(5%~10%的病例)的扩增。与3亚型髓母细胞瘤相似,这些肿瘤有数种染色体畸变,80%的病例存在等臂染色体17q。他们被分成富含等臂染色体17q的高危组(10年总生存率为36%),以及11号染色体缺失和MYCN扩增的低危组(10年总生存率为72%,这一生存率是高危组的2倍)。
髓母细胞瘤的治疗包括较大限度顺利切除,以及之后的全脑全脊髓放疗和化疗。目前的研究集中在WNT型髓母细胞瘤治疗方案的降级(目的是减轻全脑全脊髓放疗和化疗的毒性)、靶向SHH型髓母细胞瘤SMO及其下游通路的疗法,以及针对3亚型和4亚型非WNT/非SHH型髓母细胞瘤的风险调整治疗。
 胶质瘤
胶质瘤 垂体瘤
垂体瘤 脑膜瘤
脑膜瘤 脑血管瘤
脑血管瘤 听神经瘤
听神经瘤 脊索瘤
脊索瘤



 沪公网安备31010902002694号
沪公网安备31010902002694号